Contents
1 Overview
There are two different types of cgMLST schemes possible, i.e. stable and ad hoc ones. Stable schemes provide a public expandable nomenclature whereas ad hoc schemes provide a local nomenclature. Defining, evaluating, and calibrating a good stable cgMLST scheme is quite laborious. However, all approved stable schemes are publicly available and downloadable for immediate use. In contrast, users quickly do establishing an ad hoc scheme.
This tutorial describes how to use Ridom SeqSphere+ software to define, evaluate, and calibrate a stable cgMLST scheme that can be used for an expandable nomenclature. Creating a stable cgMLST scheme is a 3 step process:
- Defining the cgMLST Scheme
- Evaluating the cgMLST Scheme
- Calibrating the cgMLST Complex Type Threshold
2 Preliminaries
- Installation: This tutorial requires a running SeqSphere+ client and server. Start the SeqSphere+ server, then start the SeqSphere+ client and initialize the database. For evaluation purpose a free evaluation license can be requested.
- Tutorial Data: Download the example data archive SeqSphere_Examples_PGM_200bp_N_meningitidis.zip for this tutorial, and extract the zip-file on your computer. The example data contains Neisseria meningitidis PGM 200bp whole genome shotgun (WGS) data of 3 samples of a community outbreak that was published by U. Vogel et al. (JCM 50: 1889, 2012). Neisseria meningitidis is used exemplarily for this demonstration. However, by reading this tutorial you should be able to define your own projects for other species.
![]() Important: This tutorial is based on the genomes available from NCBI Genomes in January 2016. In later revisions of NCBI Genomes more complete genomes or chromosomes may be available and the seed genome may have a different number of annotated genes. Therefore a newly created cgMLST scheme may have a slightly different number of genes than the one described in this tutorial.
Important: This tutorial is based on the genomes available from NCBI Genomes in January 2016. In later revisions of NCBI Genomes more complete genomes or chromosomes may be available and the seed genome may have a different number of annotated genes. Therefore a newly created cgMLST scheme may have a slightly different number of genes than the one described in this tutorial.
3 Defining the cgMLST Scheme
3.1 Choosing the Seed Genome
- Each cgMLST scheme is based on a seed genome. The seed isolate/genome must fulfill the following criteria (in decreasing priority):
- The seed genome must be complete, annotated, and accessible (e.g., from NCBI). The genome should have been best done with Sanger sequencing (avoid ‘pyro’ sequenced genomes).
- The seed isolate should be available from culture collections (e.g., ATCC and/or DSMZ; sometimes difficult to fulfill for ‘biological warfare’ isolates) and DNA for sequencing must be available.
- Preferentially the seed isolate should be the type strain or another well characterized strain of the species.
- For N. meningitidis the FAM18 strain is used as seed genome, available from NCBI Genomes with accession number NC_008767
- Step 1: Choose from the menu
 Tools | cgMLST Target Definer to open the target definer dialog.
Tools | cgMLST Target Definer to open the target definer dialog.

- Step 2: Press
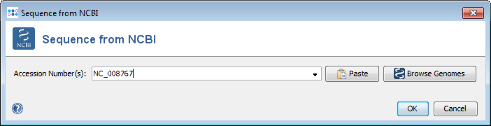
 Get from NCBI in the Seed Genome field on the top and enter the NCBI accession number of the chosen seed strain. For this tutorial this is NC_008767. Press OK to retrieve the sequence and insert it as seed genome.
Get from NCBI in the Seed Genome field on the top and enter the NCBI accession number of the chosen seed strain. For this tutorial this is NC_008767. Press OK to retrieve the sequence and insert it as seed genome.
3.2 Adding Penatration Query Genomes from NCBI Genomes
- Penatration query genomes are used to reduce the number of genes for the seed genome to a stable amount that appears in most strains of the species. It is essential that the query genomes span the whole population genetic background/variation of the species. A good beginning might be to do initially from all available finished NCBI genomes just a MLST. Subsequently, finished genomes that differ by ST and/or CC are selected as query genomes.
- Step 1: Press Add from NCBI in the Query genomes section target definer dialog.
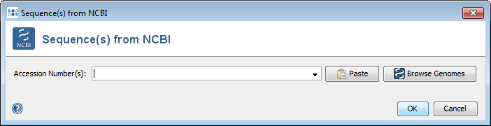
- Step 2: For this tutorial a prepared list of 13 genomes can be used. Copy the following list of NCBI accession numbers into the text field, and press OK confirm and start the download.
NC_003112, NC_003116, NC_010120, NC_013016, NC_017501,NC_017505, NC_017512, NC_017513, NC_017514, NC_017515,NC_017516, NC_017517, NC_017518

- Step 3: After the NCBI genomes are downloaded the appear in the list of Query Genomes. Do not press the Start button yet.
3.3 Adding Penetration Query Genomes from Other Sources
- If not enough NCBI complete or chromosomes bacterial genomes are available other sources can be used to search for suitable penetration query genomes:
- In the NCBI Bacteria Genome Browser window select also Scaffold and Contig as Genome Status to show the available draft genomes.
- Search at NCBI SRA, download and assemble these data and add the assembled contig FASTA files as query genomes.
- Use your own assembled sequence data as query genomes. Supported files formats are (multiple sequence) FASTA and GenBank.
3.4 Removing Outlier Genomes
- Step 1: Press the button Find taxonomic and quality outliers. Now all query genomes are compared against all seed genome genes. This process takes some while.
- Step 2: The results show that in all query genomes between 88% to 95% of the non-homologous seed genome genes were found. No obvious outlier can be identified. So all genomes can be used as query genomes.
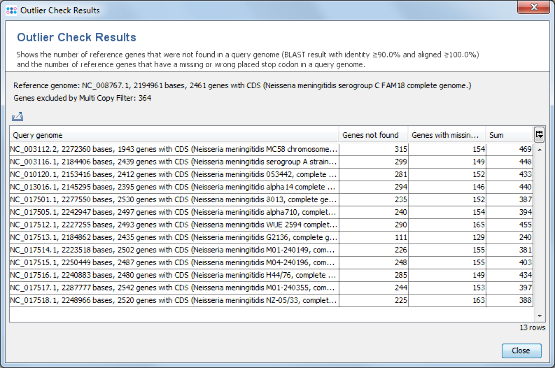
- As an aside: A good example for an outlier can be found with Pseudomonas aeruginosa. Strain PA7 (NC_009656) is described as taxonomic outlier in PLoS ONE 5 (1), E8842 (2010). This can be easily verified with the SeqSphere+ function for finding outliers:
- Open from the menu Tools | cgMLST Target Definer, and use NC_002516 as Seed Genome. Then press Add from NCBI in the Query Genomes section. Use the buttons Paste... and Paste from File to import the list of accession numbers from file P_aeruginosa_NCBI_query_genomes.txt of the example data folder. After the data is retrieved from NCBI, press the button Find taxonomic and quality outliers. The result table shows that in PA7 genome only 76% of the non-homologous genes of the seed genome were found, while in the 7 other genomes 95% to 97% of these genes were found.
3.5 Removing Plasmid Genes
- The third section in the target definer can be used to add sequence data that is used to exclude specific genes. This can be used to exclude genes that are horizontally transferred, e.g. plasmids, to prevent that such genes are part of a cgMLST typing scheme.
- Step 1: Press Add from NCBI in the Exclude Genes section of the target definer dialog.
- Step 2: Press Browse Genomes button to open again the NCBI Bacteria Genome Browser window.

- Step 3: This time Plasmids is selected in the Data Set field, and Neisseria meningitidis (the species of the seed genome) is predefined as Organism Name. Press the All button in the Genome Status panel to show all available entries. Two plasmid sequences for N. meningitidis are published only. Select both and press OK to choose them and OK to download add them to the exclude list.
3.6 Calculating the cgMLST Scheme
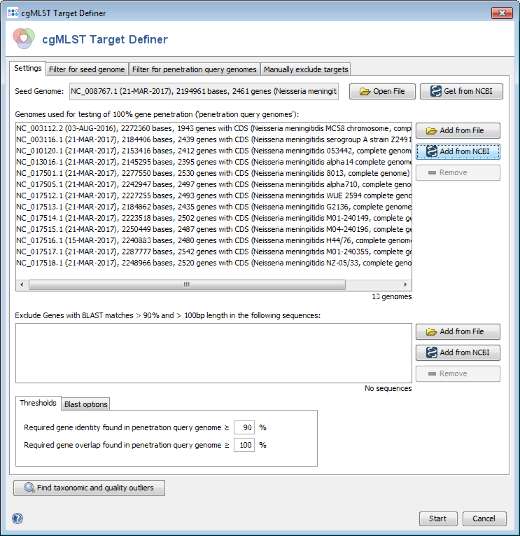
- Step 1: The cgMLST target definer dialog should now contain FAM18 as seed genome, the query genomes, and the 2 plasmid sequences. The below of the window shown settings for the thresholds and Blast options should be left to default (unless there are carefully considered reasons to change them). Press Start to initiate the cgMLST scheme definition process.
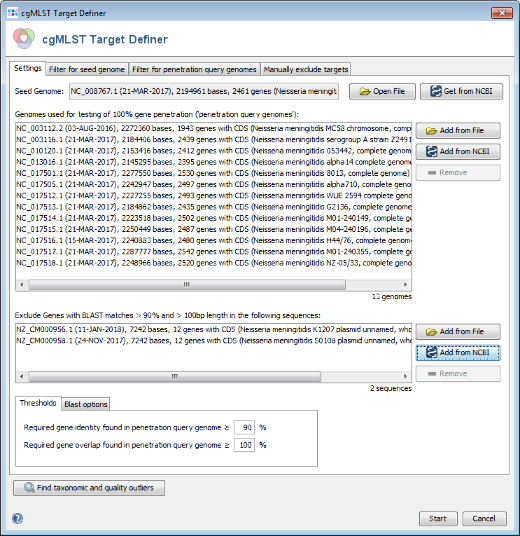
- Step 2: After a few minutes the process is finished and the results are shown. Each of the 2,461 genes of NC_008767 (21-MAR-2017) was added to one category: cgMLST, Accessory or Discarded.
- cgMLST contains 1,331 genes that are not homologous, do not have invalid start/stop codons in the seed genome, do not overlap with other genes, do appear uniquely in all query genomes with the defined thresholds, and do not have invalid stop codons in more than 80% of the query genomes. Those genes are used as targets for cgMLST.
- Accessory contains 640 genes that are not homologous and do not have invalid start/stop codons in the seed genome, but overlap with other genes, do not appear in all query genomes or have invalid stop codons in 80% or more of the query genomes. By convention, those genes are not used for cgMLST. However, they can be used in addition to increase the discriminatory power if the resolution of cgMLST is not high enough.
- Discarded contains 490 genes that are homologous or have invalid start/stop codons in the seed genome. Those genes are not used at all.
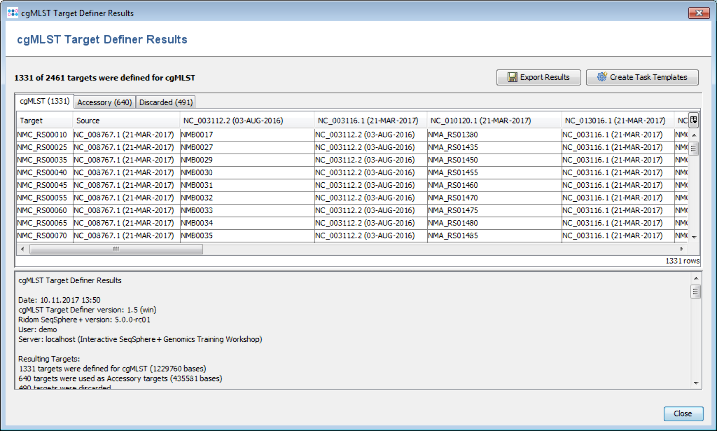
- Step 3: Press the button Create Task Templates to create two Task Templates that then can be used in a project to analyze N. meningitidis next-gen sequencing data: The cgMLST Task Template is created for the 1,331 cgMLST targets. A second Task Template called Accessory is created for the 658 genes of the Accessory category.
- Step 4: Press the Save to store the Task Templates.
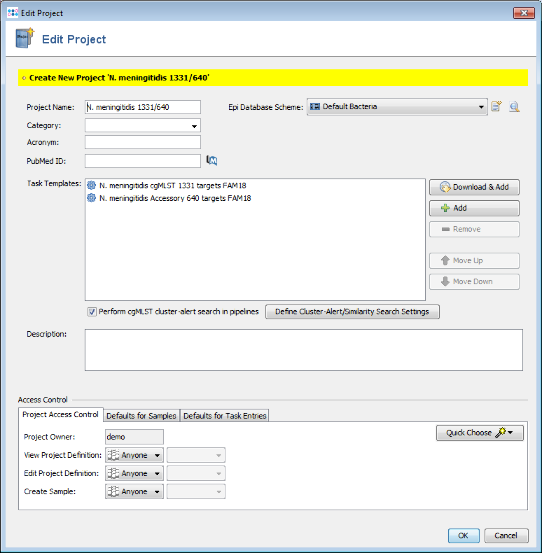
- Step 5: A new dialog is shown that allows to immediately create a Project for the new two Task Templates. Press the Yes button to open the editor for the new Project. The name of the project can be changed. Use here the default database scheme. Confirm with OK to save the new Project.
4 Evaluating the cgMLST Scheme
4.1 Evaluating with Seed Draft Genome
- As a pragmatic test for the new cgMLST scheme, the seed strain can re-sequenced. This should be done with an up-to-date NGS platform (e.g., Illumina). The read data should be either de novo assembled (e.g., Velvet or SPAdes), or reference mapped (e.g., BWA) for monomorphic/clonal bacteria.
- For the pragmatic test of the cgMLST scheme 98.5% or more of the cgMLST targets should be found and pass all automatic analysis checks. Else we strongly recommend to investigate the reason(s) for failure by running duplicates or consulting literature for targets known to be technical difficult to assemble (e.g., for MtbC PPE & PE gene family). If problems are repeatedly observed for certain targets then remove them manually from the cgMLST targets and add them to Accessory genome targets.
- This evaluation step is not done in this tutorial.
4.2 Evaluating with Diverse Isolate Collection
- For the evaluation of the cgMLST scheme a well-characterized diverse isolate collection spanning the whole population genetic background of the species should be analyzed. If it turns out that the scheme is not yet stable (some genomes have less than 95% good cgMLST targets) then acquire additional representative isolates, produce high-quality draft genomes, and add them as query genomes (iterative cgMLST scheme defining process).
- This evaluation step is not done in this tutorial.
5 Calibrating the cgMLST Complex Type Threshold
- The calibration of the cgMLST Complex Type threshold is done by retrospective analysis of well defined outbreaks and out-group isolates with identical MLST ST/MLVA/PFGE types.
- This cgMLST Complex Type calibration step is not done in this tutorial.
6 Publishing a Stable cgMLST Scheme
- Finalized and evaluated stable cgMLST schemes will be published on the cgMLST.org Nomenclature Server. Please contact us for doing so.
- Once such schemes are published, they can be downloaded as predefined Task Templates by any SeqSphere+ user. Any user of this Task Template can submit new allele types to the cgMLST.org Nomenclature Server if at least 90% of all cgMLST genes were found and basic epidemiological information about the isolate (e.g., year and country of isolation) is available.
- The publishing step is not done in this tutorial.
7 Analyzing Outbreak Data with the Stable cgMLST Scheme
- Finally the Task Templates that were created for the new cgMLST scheme can be used for a cgMLST analysis of outbreak strains.
 Hint: As long as the cgMLST scheme is not published all allele types are only locally defined.
Hint: As long as the cgMLST scheme is not published all allele types are only locally defined.
- Step 1: Choose from the menu File | Process Assembled Genome Data. The new cgMLST Project you just created and the two Task Templates should be preselected already. Now use the button
 Add from File, choose the three tutorial fasta-files, and press the Open button. Those files are de-novo assemblies (done with MIRA) of Ion Torrent data from a single community outbreak. The upcoming window optionally allows to specify among others the procedure details of these data. Just click OK to add the files to the list. Confirm again with OK to start the import process.
Add from File, choose the three tutorial fasta-files, and press the Open button. Those files are de-novo assemblies (done with MIRA) of Ion Torrent data from a single community outbreak. The upcoming window optionally allows to specify among others the procedure details of these data. Just click OK to add the files to the list. Confirm again with OK to start the import process.
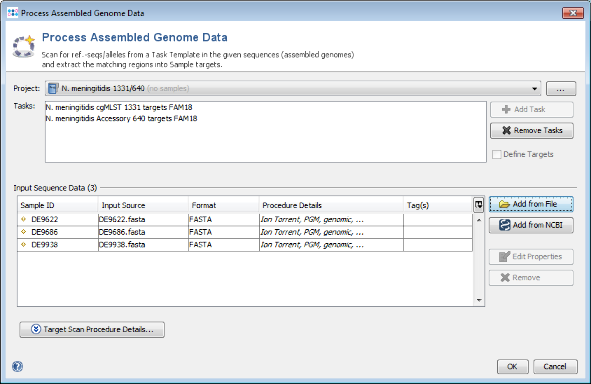
- Step 2: Ridom SeqSphere+ now loads all input sequences and tries to find (by using built-in BLAST) each of the target reference sequences that are defined in the Task Template. Once this has been done after some minutes, a dialog window opens that offers the possibility to load all three samples in the workspace. Just push the Close button of this dialog.
- Step 3: Invoke the menu command
 Tools | Comparison Table. In the Comparison Table dialog go to the first tab "Create New". In the Choose Samples section the new cgMLST project (should be preselected). In the Choose Genotypings Schemes section at the bottom cgMLST should already be preselected. Press the Create Comparison Table button to confirm.
Tools | Comparison Table. In the Comparison Table dialog go to the first tab "Create New". In the Choose Samples section the new cgMLST project (should be preselected). In the Choose Genotypings Schemes section at the bottom cgMLST should already be preselected. Press the Create Comparison Table button to confirm.
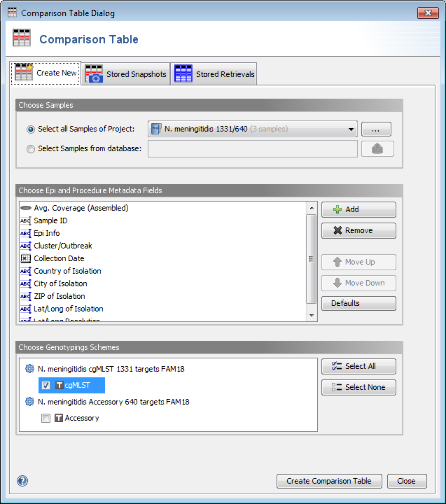
- Step 4: The comparison table is opened and shows the data for the three Samples. The columns with a green header are used for distance calculation. Table cells with red background contain missing values. Those appear if a cgMLST target was not found at all in an input sequence ("? (not found)"), if the analysis for this target has failed, e.g., because of a frame shift error ("? (failed)"), or if a new allele for this target has not yet been submitted ("? (new)").

- Step 5: Press the
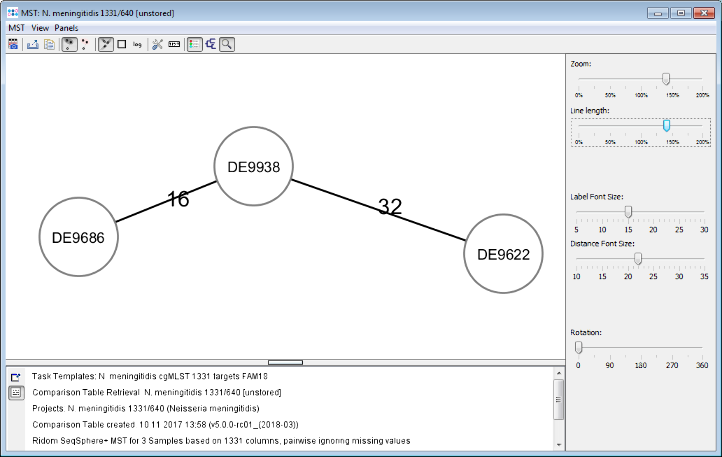
 Minimum Spanning Tree button in the toolbar to calculate the distances between the Samples and draw a minimum spanning tree for them. If the table contains missing data (targets that have no allele types assigned yet) a dialog opens. Confirm in this dialog to use the default setting, i.e. to pairwise ignore missing values in distance calculations, by pressing the OK button.
Minimum Spanning Tree button in the toolbar to calculate the distances between the Samples and draw a minimum spanning tree for them. If the table contains missing data (targets that have no allele types assigned yet) a dialog opens. Confirm in this dialog to use the default setting, i.e. to pairwise ignore missing values in distance calculations, by pressing the OK button.